While the amount of small non-coding RNA sequencing data is continuously increasing, it is still unclear to which extent small RNAs are represented in the human genome. Saarland University researchers have analyzed 303 billion sequencing reads from nearly 25,000 data sets to answer this question.
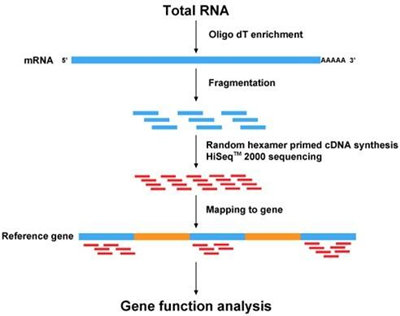
The researchers determined that 0.8% of the human genome are reliably covered by 874,123 regions with an average length of 31nt. On the basis of these regions, they found that among the known small non-coding RNA classes, microRNAs were the most prevalent. In subsequent steps, they characterized variations of miRNAs and performed a staged validation of 11,877 candidate miRNAs. Of these, many were actually expressed and significantly dysregulated in lung cancer. Selected candidates were finally validated by northern blots. While isolated miRNAs could still be present in the human genome, our presented set likely contains the largest fraction of human miRNAs.
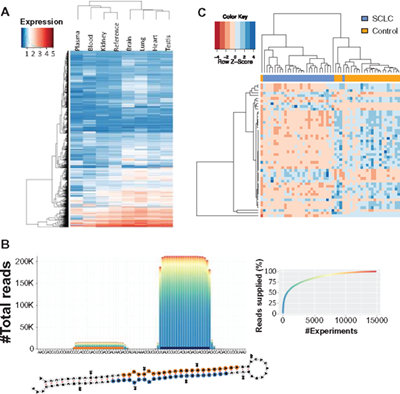
Panel (A) describes the first-pass validation of human (candidate) miRNAs by microarrays. Cluster heat map of all 4297 mature (candidate) miRNAs that showed signals in high-quality RNA samples based on amplification free hybridization. Panel (B) presents a representative example with the mapping distribution as presented in Figure 3. In addition to the pileup plot, the secondary structure is shown. Panel (C) shows the result of the human blood miRNA array hybridized with lung cancer (blue) and control samples (orange). Results are presented as heat map resulting from hierarchical clustering with dendrograms on top (clustering of samples) and at the left side (clustering of miRNAs)
标注:本文摘自互联网,已表明出处,如有侵权,请联系我司,第一时间删除。
上一篇:Single cell sequencing sheds light on why cancers form in sp
下一篇:Single cell RNA analysis identifies cellular heterogeneity a


